Illumina run setup details - (done)
- Michal Twik
- Ester Feldmesser (Unlicensed)
- Dena Leshkowitz
- Marilyn Safran (Unlicensed)
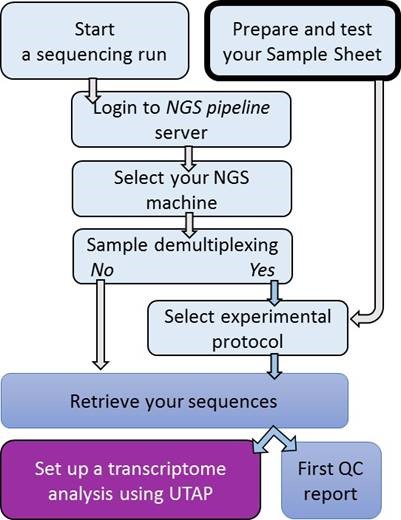
From samples to sequencing output
Note - It is your responsibility to delete the sequence data on our server. We will not store the data beyond three months. Click here to check the size of the data you currently have on storage. |
|---|
Step 1: Get a userID on SusanC
After a sequencing run starts, the user MUST login to the NGS Pipeline (SusanC3) website and register the run. This is critical for data retrieval.
To do so, you must have an account (userID and password) on the SusanC3 server. To obtain one, contact Irit Orr at irit.orr@weizmann.ac.il or 08-934-2470.
Step 2: Preparing your SampleSheet
If you request demultiplexing and quality control services, you are required to prepare a file with the details about your samples.
In the SampleSheet file you need to enter the usersIDs for the users who should receive the sequences. You may specify for selected samples to be delivered to separated users, this is demultiplexing.
The sample sheet must be prepared in a format (csv or xlsx) corresponding to one of the following three experiment protocols:
- Illumina-compatible indexing (TruSeq RNA-Seq and others) - csv file (download example) or for a protocol with 2 barcodes per sample - csv file (download example)
- Mars-Seq - xlsx file (download example)
- 10X Genomics dual index protocol (single cell RNA-Seq) - csv file (download example). Note: Leave the Lane column empty (unless you are sure that you know what you're doing...)
We recommend that you test the sample sheet format here. The same sample sheet may contain samples for several users, which should be detailed within the file.
DO NOT mix protocols on a run. Note: Valid characters for sample names are A-Z a-z 0-9 . _ - |
|---|
Mismatches (sequencing errors)
By default, we don't allow mismatches (sequencing errors).
In Illumina-compatible indexing, you can use the barcode-mismatches option, it should be added to the [Settings] in the Samplesheet, barcode-mismatches - The number of mismatches allowed per index adapter, accepted values are 0, 1, or 2. (download example)
Step 3: Once you start the run on the Illumina machine
After logging in to the NGS Pipeline (SusanC3), select "Start here to register your run"
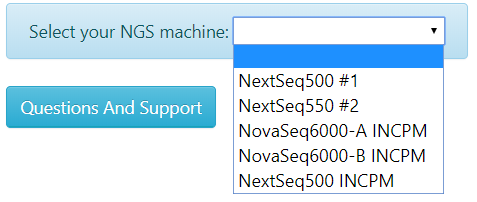
Select your sequencing machine:
| Warning: Use NovaSeq with unique dual indexing pooling combinations (unique i5 and i7 indexes). Index hopping or index switching is a known phenomenon in NovaSeq. It causes incorrect assignment of libraries from the expected index to a different index (in the multiplexed pool). For some protocols, e.g. 10X Genomics, scRNA-Seq index hopping contaminations can be eliminated using bioinformatics procedures. Please consult with us at utap@weizmann.ac.il. |
|---|
As shown below, your logged-in user name and email address are filled in automatically by the system in the User field.
Please provide the userid of of the Principal Investigator in the PI userid field, and click "Next step":

Additional services for sequences
In order to use post-sequencing services, such as demultiplexing and quality control, please select "Yes" as shown below:

If you request demultiplexing and quality control services, you are required to upload a sample sheet to the NGS Pipeline server at least 5 minutes after a NextSeq machine has begun running, but before sequencing has completed.
Note that a sequencing run typically takes a few hours or more to complete, depending on the number of samples.
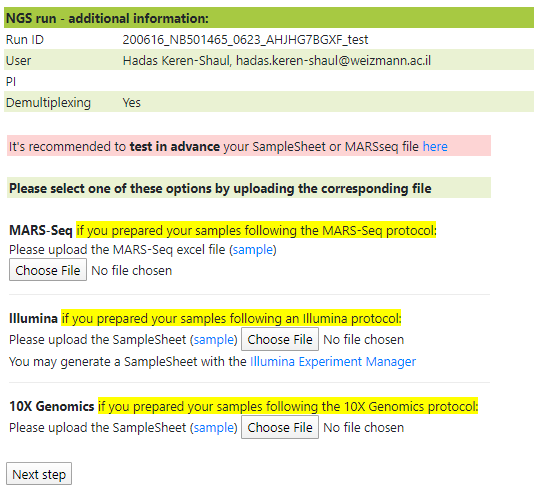
The sample sheet will be analyzed, Make sure to review the sample names before submission:
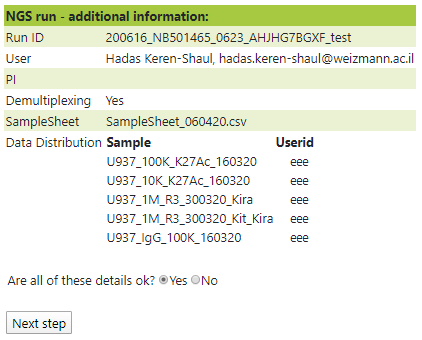
If a sample sheet is uploaded in the correct format, demultiplexing and quality control will be executed automatically.
Raw (Bcl and Fastq) output files from the sequencing machine are temporarily stored on the Stefan server (same userID and password as on SusanC3). Upon sequencing completion and automatic transfer to the Stefan server, you may choose to simply download raw fastq files from the Stefan server (created by the sequencing machines). Instructions are available at Getting your sequencing data.
Email notification
An email will be sent to you upon sequencing completion, with instructions about downloading Bcl and/or Fastq files. If you selected post-sequencing services, the files will be demultiplexed, and an additional link for viewing QC results will be included in the email (using the template http://stefan.weizmann.ac.il/fqc/<RUN_ID>).
Data Backup and Deletion
Please back up your files and delete them from the Stefan server using the storage service. Note that your files will be automatically deleted 3 months after they are created.
List of links
Sequencing pipeline:
https://ngs-pipeline.weizmann.ac.il/
Help page:
https://ngs-pipeline.weizmann.ac.il/ngsb/howto
QC:
http://stefan.weizmann.ac.il/fqc/{type RUN ID here}, for example:
http://stefan.weizmann.ac.il/raw/180509_NB551168_0120_AHTKN2BGX5/
List of runs, with delete functionality:
https://ngs-pipeline.weizmann.ac.il/ngsb/storage
Bioinformatics support staff:
- Bioinformatics team biopipe@weizmann.ac.il
- UTAP team (UTAP maintenance) utap@weizmann.ac.il
- Dena Leshkowitz
- Ester Feldmesser
- Gil Stelzer
- Bareket Dassa
- Noa Wigoda