UTAP: ATAC-seq pipeline guidelines
The ATAC-seq (Assay for transposase-accessible chromatin using sequencing) pipeline facilitates the analysis of ATAC-seq data in order to capture open and accessible regions of chromatin across the genome. The pipeline receives paired-end reads as input, performs quality control and pre-processing steps, and maps the reads onto mouse or human genomes. Nucleosome-free fragments are selected (after some post-processing), and peaks identified and analyzed.
This pipeline is available in https://utap.wexac.weizmann.ac.il/
Before you start:
This pipeline runs on the Wexac cluster.
Please prepare the following in advance:
- An account (userID) on Wexac, via your department administrator.
- A "Collaboration" folder within your lab folder on Wexac, with read and write permission for Bioinformatics Unit staff. This must be set up by the computing center (hpc@weizmann.ac.il).
- Sufficient free storage space on Wexac (> 400Gb), via your department administrator.
In order to run a new ATAC-seq analysis, you must first transfer demultiplexed sequencing data (fastq files) to your Collaboration folder. Within the Collaboration folder, a directory structure will be created with outputs of the analysis described below.
Setting up a new analysis
If you wish to run a new analysis from existing files in the Collaboration folder, or if you’ve uploaded new sequencing data (not produced within the LSCF) to Wexac from an external source, login to UTAP (ngsbio.wexac.weizmann.ac.il) via Firefox or Chrome (the pipeline is NOT compatible with Internet Explorer) using your Weizmann userID and password.
Click "Run pipeline"
- Select the type of analysis you desire:
- Select the input folder:
Browse within your Collaboration folder and select the folder containing your sample files (fastq). Fastq files must be organized, within the selected folder (root folder), into subfolders as shown below.
Note that if you wish to go up one level (or more), click the desired folder level on the path at the top of the window.
Fastq file name conventions: Fastq file names must start with the same sample name as the subfolders, and end with "_R1.fastq" (or "_R1.fastq.gz") for single-read data . In the case of paired-end data, corresponding files must exist that are IDENTICAL in their name, but contain the suffix "_R2.fastq" (or "_R2.fastq.gz") instead of "_R1.fastq", where R is the read number.
For example:
- root_folder
- sample1
- sample1_R1.fastq
- sample1_R2.fastq (must exist in paired-end)
Or:
- sample2_R1.fastq.gz
- sample2_R2.fastq.gz (must exist in paired-end)
The pipeline also supports the fastq file format conventions _S*_L00*_R1.fastq or _S*_L00*_R1_0*.fastq.
For example:
- root_folder
- sample1
- sample1_S0_L001_R1_001.fastq
- sample1_S0_L001_R1_002.fastq
- sample1_S0_L002_R1_001.fastq
- sample1_S0_L002_R1_002.fastq
- sample1_S0_L001_R2_001.fastq (must exist in paired-end)
- sample1_S0_L001_R2_002.fastq (must exist in paired-end)
- sample1_S0_L002_R2_001.fastq (must exist in paired-end)
- sample1_S0_L002_R2_002.fastq (must exist in paired-end)
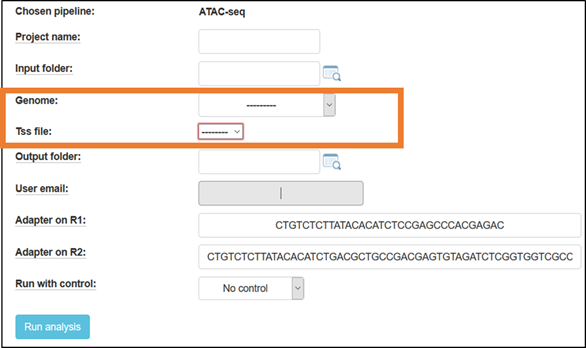
UTAP user interface input information required:
- Select the output folder
If you want the output folder to be different from the one automatically filled in (based on the selected input folder), overwrite the output folder name in the text box associated with the screen’s Output folder: field with your name of choice.
- Additional setups
Fill in a project name, and select the reference genome to which the reads will be aligned.
For mouse, you have the option to choose a TSS file, containing either a broad or narrow definition of the genes’ TSS (Transcription Start Site) regions (based on Nature. 2016 Jun 30;534(7609):652-7 - The landscape of accessible chromatin in mammalian preimplantation embryos).
Default adapters are P5 and P7 adapters of the Tru-seq protocol.
By default, the ATAC-seq pipeline runs without a “black-list” or “naked-DNA” control.
- Run the pipeline
Finally, click the “Run analysis” button to submit the analysis. Once the analysis is completed, you will be notified by email (usually after a few hours).
All of the output files will be stored in your Wexac Collaboration folder.
At this point no report is being created.
Analysis workflow
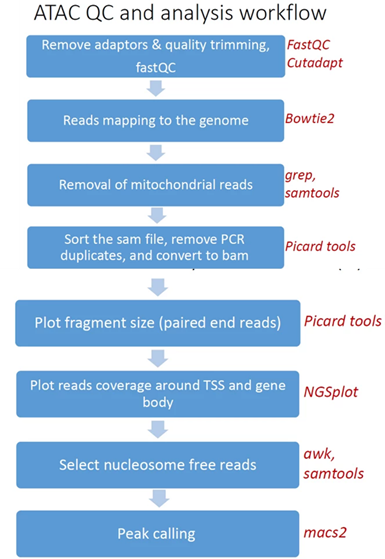
Pipeline steps and associated tools:
- Reads trimming: Reads are quality trimmed using cutadapt. In this process primers corresponding to the TruSeq protocol are removed (output is in folder 1).
- Quality control: Reads quality control is evaluated using FastQC (in output folder 2), and a report file, containing quality reports for all of the samples, is generated using multiQC (in output folder 3).
- Mapping to genome: The quality trimmed paired-end reads are mapped to Mouse/Human genomes using Bowtie2 (output is in folder 4).
- Alignment filtering: Following the alignment, mitochondrial genes are removed from the analysis (using the grep command). Duplicated reads are removed using picard-tools. The remaining unique reads are indexed and sorted using samtools index and samtools sort. Statistics on the alignment is generated using flagstat (output is in folder 5).
- Select nucleosome-free fragments: fragments of length <120bp are selected using the awk command (alignments are in folder 6), and insert size distributions are plotted before and after size selection (output is in folder 8, plots after selection end with "_nucl_free").
- Visualization in graphs: reads coverage on gene body and around the TSS are graphically visualized using ngsplot (output is in folder 7).
- Read counts on TSS: for mm10 genome we count the number of reads on genes’ TSS (Transcription Start Site) regions based on, Nature. 2016 Jun 30;534(7609):652-7 ).
- Peak calling: Broad peaks are called using MACS2 (output is in folder 10).
Output folders:
1_cutadapt
2_fastqc
3_multiqc
4_mapping
5_process_alignment
6_nucleosome_free
7_ngs_plot
8_picard_plot
9_tss_count
10_call_peak
11_reports
Log files (one directory above the output directory):
snakemake_stdout.txt