16.2.22 UTAP: Ribo-Seq Pipeline
Pipeline to run Ribo-Seq analysis from sequences to UTR and CDS counts and MACS peaks to determine ribosome enrichment regions.
Developed by Dena Leshkowitz and programmed for the UTAP pipeline by Michal Twik and Jordana Lindner. Details regarding the pipeline are available here.
Pipeline website: http://utap.wexac.weizmann.ac.il
Setting up a new analysis
The transcriptome pipelines run on the Wexac cluster. In order to run a new transcriptome analysis, your fastq files must be in your Collaboration folder on Wexac, in the correct structure (UTAP requirements & description). Then, login to utap.wexac.weizmann.ac.il via Firefox or Chrome (the pipeline is NOT compatible with Internet Explorer) using your Weizmann userID and password, and click on Run pipeline:
Click on "Ribo-Seq" in the Choose pipeline box
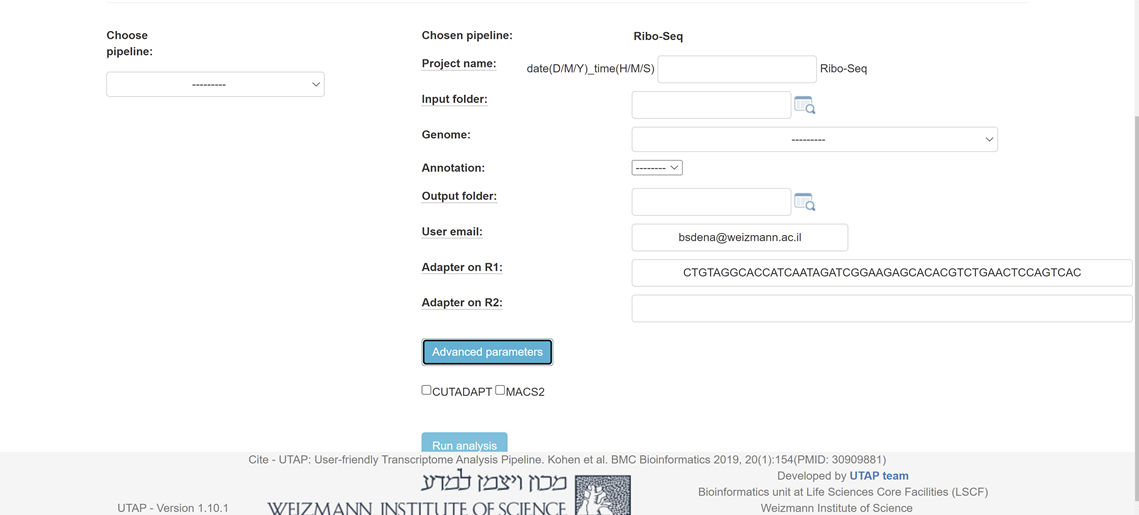
Please fill all the relevant information in the "choice box" .
LINK:
Transcriptome pipeline for Weizmann Institute users: http://utap.wexac.weizmann.ac.il
Descriptions of analysis of ribosome profiling sequences:
Initial steps are similar to described in Ingolia et al. Nature 2012
Removing first base and adapter and keeping only sequences of length 20 to 50 (initial length 61) that had adapter, not trimming by quality.
cutadapt -a CTGTAGGCACCATCAATAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC --discard-untrimmed --times 1 -u 1 -m 20 -M 50
rRNA removal
Database of rRNA was downloaded from http://genome.ucsc.edu/cgi-bin/hgTables
Then set group: all tables, table: rmsk, and filter to "repClass (does match) rRNA"
Run bowtie to make fastq file of non-RNA sequences
bowtie -p 10 -norc --un CHX_D101_S1_L003_R1_001_no_rRNA_cutadapt.fastq ../rRNA/rRNA
Filtering for reads of length 28-33 using cutadapt (-m 28 -M 33)
Mapping the reads to mm10 mouse (or to hg38 genome) using TopHat2 with parameters : --no-novel-juncs --library-type fr-firststrand
Bam files were converted to tdf files using igvtools count, in order to view with IGV.
Quantifying reads on 5’-UTRs and CDS
UTR and CDS gtf files were created from refseq gtf file, avoiding regions found in both files. HTSeq was used for the quantification (parameters -s yes -t exon -m intersection-nonempty). The option intersection-nonempty allows to assign junction reads to either the CDS or UTR, depending where most of read is placed.
Finding Ribosome profiling summits
Peaks were identified using macs (-g 3e9 --keep-dup all --nomodel --shiftsize=1) with no model and no filtering of PCR duplicates.
Summit peaks were annotated by intersection (intersectBed) with refseq gtf file, shifted 12 bases and expanded to 3 bases (awk commands).
Acknowledgments
Citation:
Kohen et al. BMC Bioinformatics (2019) 20:154 https://doi.org/10.1186/s12859-019-2728-2 (PMID: 30909881)
Bioinformatics support staff for UTAP:
- UTAP development and maintenance team: utap@weizmann.ac.il