UTAP guidelines- User-friendly Transcriptome Analysis Pipeline
- Bareket Dassa
- Dena Leshkowitz
- Jordana (or admin)
Pipeline website: https://utap.wexac.weizmann.ac.il/
Information on 16.2.22 UTAP version history
A more comprehensive summary and guide to installing the system on your local cluster: https://utap.readthedocs.io
A guide for ChIP-seq and ATAC-seq pipelines: UTAP: ChIP-seq pipeline guidelines, UTAP: ATAC-seq pipeline guidelines
Login using your Weizmann userID and password.
UTAP is an intuitive and scalable transcriptome pipeline, which enables fast and user-friendly data analysis. The pipeline executes the full process, starting from sequences (RNA-Seq and bulk Mars-seq) and ending with sets of differentially expressed genes. Output files are placed in a structured folder system, and summarization of the results is displayed in a rich and comprehensive report.
Before you start:
In order to run a transcriptome analysis please prepare in advance:
- An account (userID) on Wexac (may be setup by your department administrator)
- A "Collaboration" folder with read and write permissions for the Bioinformatics within your lab folder. This must be setup by the computing center (hpc@weizmann.ac.il).
- Sufficient free storage space on Wexac (> 400Gb). May be setup by your department administrator.
The transcriptome pipelines run on the Wexac cluster. In order to run a new transcriptome analysis you must first transfer demultiplexed sequencing data (fastq files) to your Collaboration folder on Wexac. Within the Collaboration folder, a directory structure will be created according to the transcriptome analysis setup.
Setting up a new analysis
If you wish to run a new analysis from existing files in the Collaboration folder, or you uploaded data to Wexac from an external source (sequencing data not performed in the LSCF), login to ngsbio.wexac.weizmann.ac.il using your Weizmann userID and password.
The pipeline is compatible with Firefox and Chrome browser, but not with InternetExplorer.
Click "Run pipeline"
1. Select the type of analysis you desire:
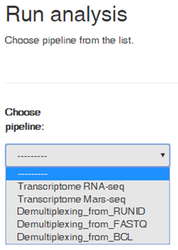
2. Select the input folder:
Browse within your Collaboration folder and select the folder containing your sample files (fastq). Fastq files must be organized, within the selected folder (root folder), into subfolders as shown below.
Note that if you wish to go up one level (or more) please click the desired folder level on the path at the top of the window. See below the Fastq file name convention.

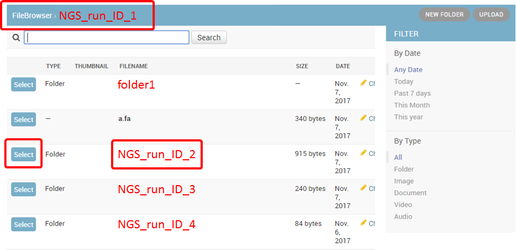
Fastq file names must start with the same sample name as the subfolders and end with "_R1.fastq" (or "_R1.fastq.gz") for single-read data . In the case of paired-end data (required for Mars-Seq), corresponding files must exist that are IDENTICAL in their name except for the ending "_R2.fastq" (or "_R2.fastq.gz") instead of "_R1.fastq".
Where R is the read number.
For example:
- root_folder
- sample1
- sample1_R1.fastq
- sample1_R2.fastq (must exists in Mars-seq and in paired-end)
- sample2
- sample2_R1.fastq.gz
- sample2_R2.fastq.gz (must exists in Mars-seq and in paired-end)
- sample1
The pipeline also support the convention of the fastq file format _S*_L00*_R1.fastq or _S*_L00*_R1_0*.fastq.
For example:
- root_folder
- sample1
- sample1_S0_L001_R1_001.fastq
- sample1_S0_L001_R1_002.fastq
- sample1_S0_L002_R1_001.fastq
- sample1_S0_L002_R1_002.fastq
- sample1_S0_L001_R2_001.fastq (must exists in Mars-seq and in paired-end)
- sample1_S0_L001_R2_002.fastq (must exists in Mars-seq and in paired-end)
- sample1_S0_L002_R2_001.fastq (must exists in Mars-seq and in paired-end)
- sample1_S0_L002_R2_002.fastq (must exists in Mars-seq and in paired-end)
- sample2
- sample2_S0_L001_R1_001.fastq
- sample2_S0_L001_R1_002.fastq
- sample2_S0_L002_R1_001.fastq
- sample2_S0_L002_R1_002.fastq
- sample2_S0_L001_R2_001.fastq (must exists in Mars-seq and in paired-end)
- sample2_S0_L001_R2_002.fastq (must exists in Mars-seq and in paired-end)
- sample2_S0_L002_R2_001.fastq (must exists in Mars-seq and in paired-end)
- sample2_S0_L002_R2_002.fastq (must exists in Mars-seq and in paired-end)
- sample1
3. Select the output folder
If you want the output folder to be different from the one automatically filled in (based on the selected input folder), select the desired output folder.
4. Additional set ups:
Fill in a project name, select the reference genome and annotation for which the reads will be aligned to.
Select if your protocol is stranded (the sequenced reads saves the original strand of RNA fragments) or non-stranded. If you don't know, select in "find automatically" option.
Type your adapters on each read (R1 and R2). These adapters will be removed from the reads by the pipeline. You can remain the default adapters if you use with P5 and P7 adapters of True-seq protocol).
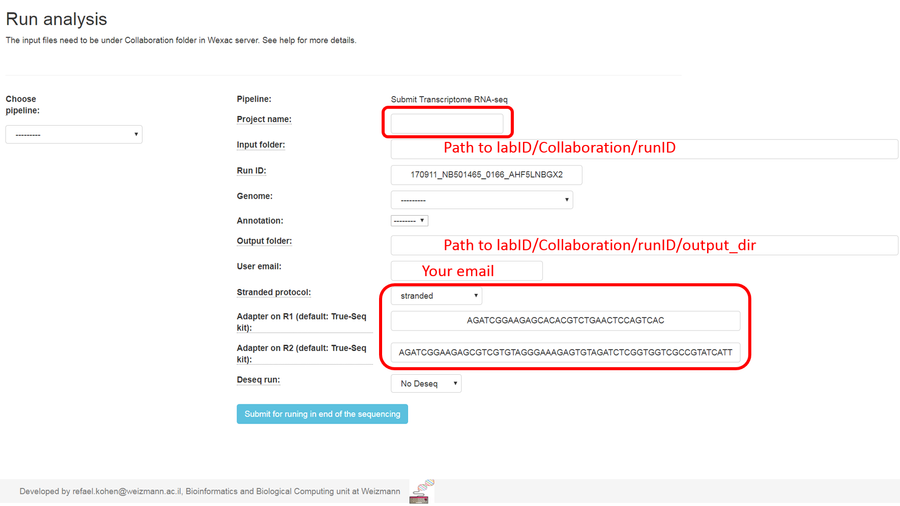
5. Differential gene expression analysis
Choosing “run DESeq2” allows you to detect differentially expressed genes in your data using the DESeq2 package (DESeq2 manual). If you selected this option, by default, at least two categories must be created.
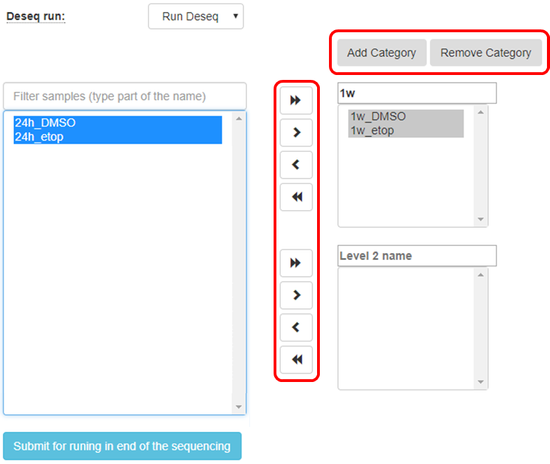
Create categories for the treatments that you would like to compare, fill in the category names, and sort the samples accordingly as shown in the figure below:
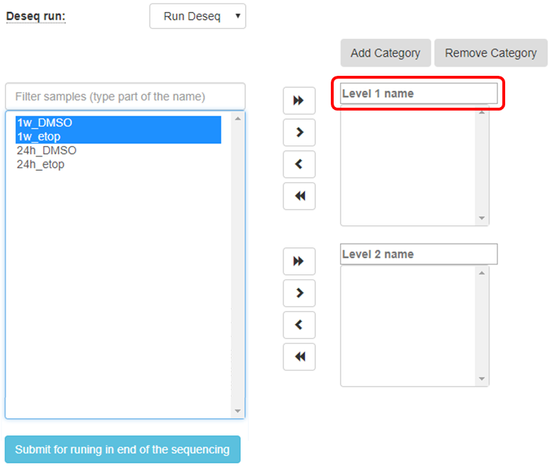
Sort the samples by selecting them and using the arrows to move to the appropriate categories
You may add additional categories by using the proper buttons:
If the samples were prepared in different batches, you can add this information: After the moving the samples into categories boxes, click on "Add Batch Effect" button, then select the samples that belongs to one batch and click on "Batch 1" button.
Return on the operation with the other groups of the samples.
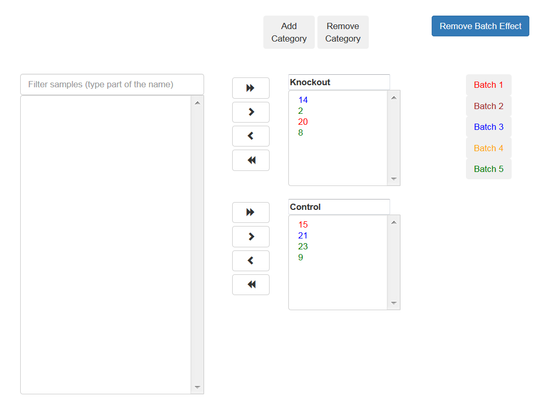
All steps of the pipeline (mapping, counts etc.) will be run on all samples, but Deseq will be run only on the samples with categories.
6. Run the pipeline
Finally, submit the run for analysis. Once the analysis is completed, you will be notified by email (usually lasts a few hours). All output files will be stored on your Wexac Collaboration folder.
Analysis pipeline steps and reports
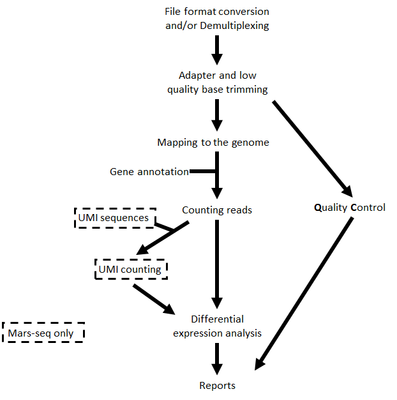
The steps performed by the pipeline -
- Trim adapter sequences
- Fastqc for quality control of the samples will be run in parallel to the steps described
- Map reads to the selected reference genome
- Add UMI and gene information to the reads
- Quantify gene expression by counting reads
- Count UMI's to aviod PCR duplications
- Detect Deferentially Expressed (DE) genes for a model with a single factor
Steps 4 and 6 are performed only for Mars-Seq
Steps 6 is performed only if DESeq2 is selected
Upon completion, you will get an email with links to the results report
The report includes several sections -
- Sequencing and Mapping QC
- Figure 1 - Plots the average quality of each base across all reads. Quality of 30 (predicted error rate 1:1000) and up is good
- Figure 2 - Histogram showing the number of reads for each sample in raw data
- Figure 3 - Histogram showing the percentof reads discarded after trimming the adapters (after the removing of the adapters some read and polyA/T or low quality reads may be too short and the pipeline discards them)
- Figure 4 - Histogram with the number of reads for each sample in each step of the pipeline
- Figure 5 - Plots sequence coverage on and near gene regions
- Figure 6 -
- Histogram showing the percent of reads that mapped uniquely and not uniquely per sample
- Histogram showing the percent of the uniquely mapped reads that mapped to genes (genes included must have at least 5 reads)
- Exploratory Analysis
- Figure 7 - Heatmap plotting the highly-expressed genes (above 5% of total expression). For example the expression of gene RN45S in sample SRR3112243 amounts to 15% of the expression
- Figure 8 - Heatmap of Pearson correlation between samples according to the gene expression values
- Figure 9 - Clustering dendogram of the samples according to the gene expression
- Figure 10 - PCA analysis
- Histogram of % explained variability for each PC component
- PCA plot of PC1 vs PC2 c. PCA plot of PC1 vs PC3
- Differential Expression Analysis (this section exists only if you run the DESeq2 analysis) - a table with the number of differential expressed genes (DE) in each category (up/down) for the different contrasts. In addition, links for p-value distribution, volcano plots and heatmaps as well as a table of the DE genes with dot plots of their expression values
- Bioinformatics Pipeline Methods - description of pipeline methods utilized
- Links to additional results - links for downloading tables with raw, normalized counts, log normalized values (rld) and statistical data of contrasts. In case of model with batches, "combat" values were calculated (instead of rld) using "sva" package and are batch corrected normalized log2 count values.
Annotation file:
For the counts of the reads per gene we use with annotation files (gtf format) from "ensemble" or "gencode". In MARS-seq analysis we use a window within the transcript, that contains an extended 3' UTR exon. It is extended and into the intergenic region (100 bases downstream from TSS) and from 3' UTR exon towards the 5' direction on the mRNA (up to 1000 bases).
Examples of reports
RNA-Seq example: public data set from Klepikova AV et al. BMC Genomics. 2015 Jun 18;16:466
https://bip.weizmann.ac.il/rna-seq
Mars-seq example: public data set from Feigelson SW et al. Cell Rep. 2018 Jan 23;22(4):849-859
https://bip.weizmann.ac.il/mars-seq
Please regard this analysis as a good starting point and not an end result.
Additional links
Information regarding Next-seq set up, reports and deleting runs:
- Help page to set up information for sequencing on Sandbox Next-Seq machines : http://susanc.weizmann.ac.il/ngs/howto.html
- List of runs and deleting runs: http://susanc.weizmann.ac.il/ngsb/storage
- Sequence QC Reports generated after run is completed: http://stefan.weizmann.ac.il/fqc/{type RUN ID here
For example: http://stefan.weizmann.ac.il/fqc/171019_NB501465_0188_AHHL7TBGX3
Information regarding UTAP pipeline:
- Directions on how to run UTAP pipeline directly after sequencing in SandBox unit: From samples to analyzed NGS data & UTAP
Transcriptome pipeline: https://utap.wexac.weizmann.ac.il/
Demo UTAP interface: UTAP - User-friendly Transcriptome Analysis Pipeline (for external users)
- Pipelines for demultiplexing: Additional NGS pipelines
- Run pipelines from command line:
UTAP pipeline for MAR-Seq - run with command line
UTAP pipeline for RNA-Seq - run with command line
Acknowledgment
Programming
- Refael Kohen
NGS analysis
- Dena Leshkowitz
- Ester Feldmesser
- Gil Stezler
- Bareket Dassa
System support
- Kiril Kogan
Initial scripts used in the pipeline were provided by INCPM Bioinformatics Unit