16.2.22 UTAP: DESeq2 from counts matrix
Pipeline to perform DESeq2 analysis from a matrix containing raw counts per gene per sample, in at least two conditions, with at least two replicates per condition.
Pipeline website: http://utap.wexac.weizmann.ac.il
Setting up a new analysis
The transcriptome pipelines run on the Wexac cluster. In order to run a new transcriptome analysis, your fastq files must be in your Collaboration folder on Wexac, in the correct structure (UTAP requirements & description) .
Before running the pipeline, prepare your counts matrix file and transfer it to the Collaboration folder.
Counts matrix file format and structure:
- The counts matrix file should be in csv or txt (tab delimited file) format.
- The matrix should contain the genes as rows (each row is a gene) and the samples as columns (each column, other than the first, is a sample). The first column should contain the gene symbols.
- When creating the counts matrix in excel, make sure to initially format the cells in the genes column to be Text (right-click on the column and select Format cells) to ensure that all gene symbols, including those that look like dates, retain the names that you enter. Unfortunately, the default General format in excel causes gene symbols that look like dates (e.g. SEPT6, MARCH5) to be treated as customized dates (with format d-mmm - 6-Sep, 5-Mar for our examples), and sometimes produces duplicated names. (Note that subsequently changing the format to Text does NOT bring back the names that were originally entered; instead, they are converted to numbers!). HGNC has committed to changing such names but it might take some time. For explanations on how to manipulate excel files see EXCEL tips.
Then, login to utap.wexac.weizmann.ac.il via Firefox or Chrome (the pipeline is NOT compatible with Internet Explorer) using your Weizmann userID and password, and click on Run pipeline:

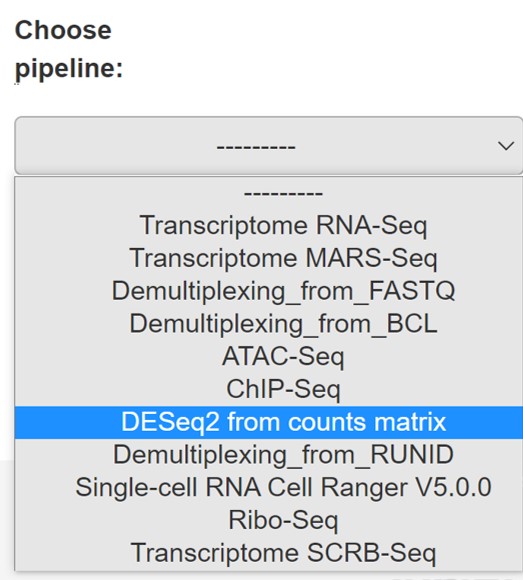
Click on "DESeq2_from counts matrix" in the Choose pipeline box
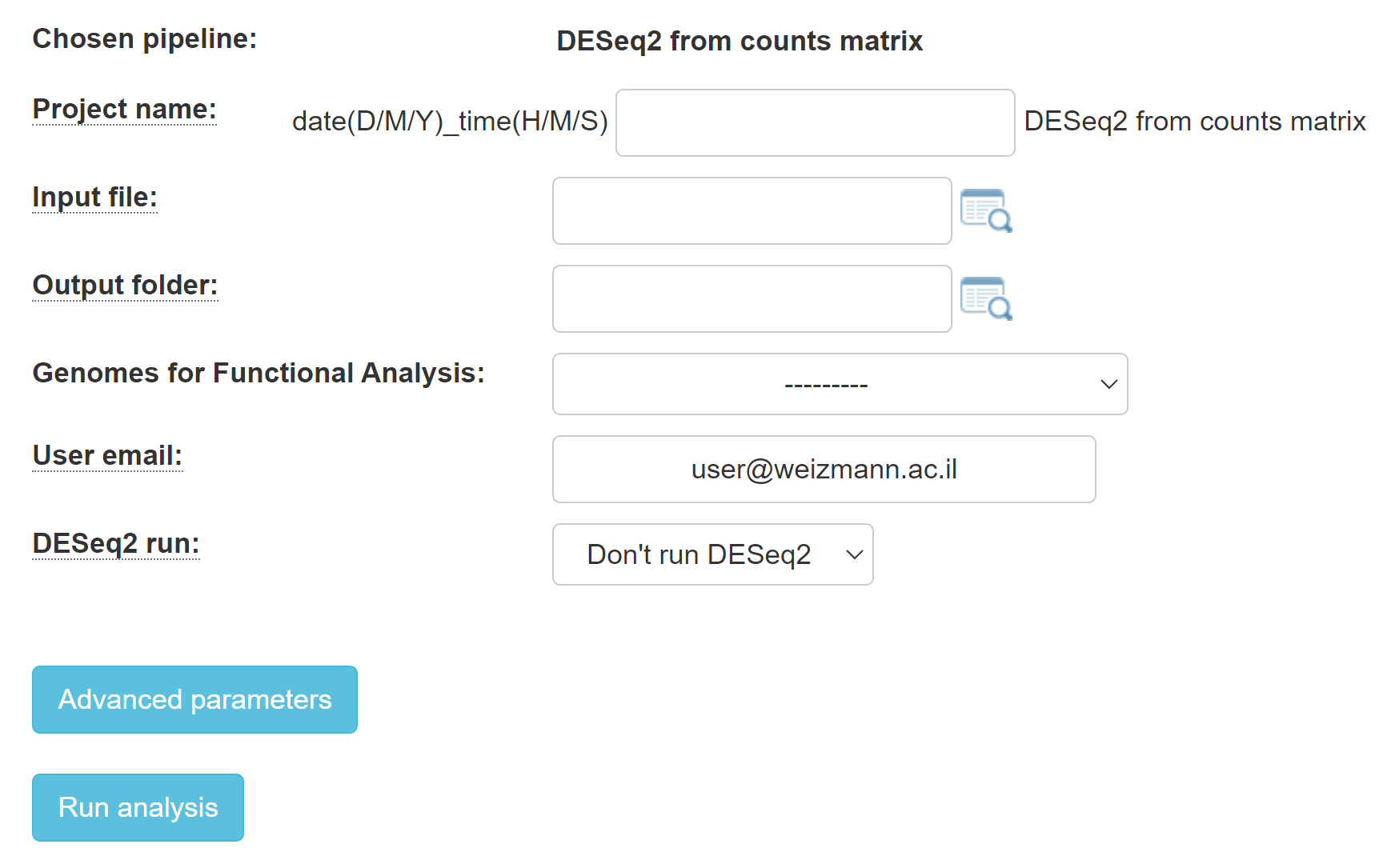
Choose your counts matrix file using 'Input folder' and click on run DESeq2 in order to identify differentially expressed genes with the DESeq2 package as described in the DESeq2 manual.
All of the samples from the counts matrix file will be parsed to the popup "choice box" .
Fill in you desired report folder name in the relevant field.
When choosing to run DESeq2 (with the 'DESeq2 run'), at least two categories must be created (by filling in the category names and dragging the relevant samples). Additional explanations can be found in 16.2.22 UTAP: Transcriptome from RNA-Seq, MARS-Seq or SCRB-Seq.
LINK:
Transcriptome pipeline for Weizmann Institute users: http://utap.wexac.weizmann.ac.il
Acknowledgments
Citation:
Kohen et al. BMC Bioinformatics (2019) 20:154 https://doi.org/10.1186/s12859-019-2728-2 (PMID: 30909881)
Bioinformatics support staff for UTAP:
- UTAP development and maintenance team: utap@weizmann.ac.il
- Dena Leshkowitz
- Ester Feldmesser
- Gil Stelzer
- Bareket Dassa
- Noa Wigoda